
Unraveling the mystery: How FDA regulations kept COVID tests out of reach just a year ago
FDA red tape hindered COVID test availability. It is a familiar narrative that echoes the challenges faced by the first-ever pregnancy tests in the early 1970s.

I will never forget where I spent the day before Christmas 2021. I was standing in line with my girlfriend, waiting for a COVID test, frozen by a Mid-Atlantic winter.
Earlier in the morning, I felt a scratchy throat, so naturally, I went searching for a test. After calling countless doctor's offices and drug stores and coming up empty, we finally found a testing center an hour away that could take us. But we would have to wait in line.

After standing out in the cold for hours, we each got two tests, a rapid test and a PCR. The rapid test came back negative in 15 minutes or so. As for the PCR tests, we never got them back.
The entire process was a complete mess.
And we weren't alone. All across the United States, tests were hard to find. But if you cracked open a newspaper, you could read stories of the Europeans having no problem finding them. The COVID wave hit them just as hard, but they seemed to weather it much better. Germany, in particular, was a standout.
Like many of my other niche fascinations, I became obsessed: Why were rapid tests so hard to find a little over a year ago?
In between projects last year, I worked on this question. And a couple of weeks back, my findings were published over at the Center for Growth and Opportunity at Utah State University.
This Twitter thread has all the important finds from the paper.
Like others that have looked into the shortages, I found that the Food and Drug Administration (FDA) bears much of the blame for limited rapid tests. But I was surprised that the same story had been repeated every decade since the early 1970s.
Back in 1971, Faraday Labs’ Ova II pregnancy test hit the market, becoming the first direct-to-consumer pregnancy test. But the test wasn’t available for long because, in December 1972, the FDA recalled it, saying that they were “inaccurate, unreliable and prone to give false results.”
Funny enough, the agency still allowed the same tests to be processed in labs. An op-ed by one FDA official was telling because he said that women “do not in general have sufficient training to detect malfunctions.” So in effect, the agency claimed that the kit was only inaccurate because it was in the hands of “laywomen,” who were prone to misreading the results. Later research would confirm that error rates were about the same for labs as for the women taking the tests.
Fast forward a half-century and the same demand for accuracy persisted for rapid tests. The US was the only developed country that required an additional set of trials that proved rapid tests were “usable.” Separate from the clinical study establishing the accuracy of the tests, the FDA required that tests undergo a usability study to prove that the general public can use them.
Usability studies and a bevy of other requirements added serious costs to US rapid tests. The real proof can be found in the prices between us and Germany, which are laid out below. German tests were cheap.

There’s so much more in the paper. It explores the economic incentives behind delays and the legal framework of diagnostic testing regulation. It also does a much better job of explaining why the US and Germany are different. Ultimately, I think the paper makes a strong case for reform.
So what can be done? There are several possible reforms that Congress will need to take the lead. They should:
Amend the Emergency Use Authorization (EUA) process to consider the cost of time;
Create clear reporting requirements on EUA approvals;
Rethink the need for diagnostic usability trials;
Refocus the standards for clinical trials for diagnostics;
Formalize rules for diagnostics; and
Mandate that the agency produce an official report on the costs of decision delays during COVID.
The FDA needs reform because there is clear evidence of regulatory bias. As Joseph A. DiMasi, Christopher-Paul Milne, and Alex Tabarrok first outlined, FDA divisions have widely varying approval times for drug approvals. Oncology drugs and antivirals get approved much faster than other drugs in neurology and cardiovascular. Truth be told, when I first saw them, the charts from the paper shocked me.
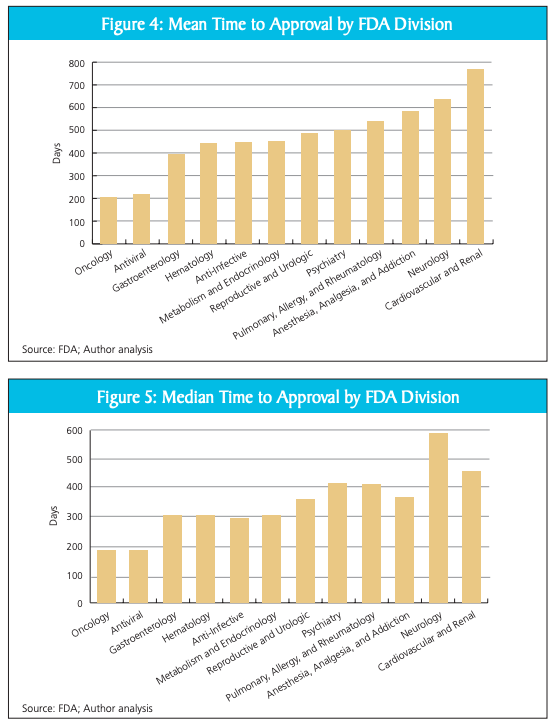
During emergencies, the FDA ought to expedite its processes without compromising safety in bringing diagnostics to market. Although the modifications I’ve laid out in the paper may not entirely resolve indecision, they could significantly contribute towards ensuring that future diagnostics are made available to individuals at the most crucial moments.
But testing isn’t the only place where the FDA needs reform. The agency needs to rethink its method of approving drugs. In upcoming work, I hope to explore what’s happening, what’s at stake, and how to change up the process.
As always, thanks for reading!
The paper can be found here, and a Twitter thread can be found here. Retweets are always appreciated.